Calvin, W. H. (1980). Normal repetitive firing and its
pathophysiology. In: J. Lockard and A. A. Ward, Jr (eds) Epilepsy:
A Window to Brain Mechanisms. Raven Press. New York.
97-121.
Copyright 1980 by author and publisher
Normal Repetitive Firing and Its Pathophysiology
William H. Calvin
Department of Neurological Surgery, University of Washington, Seattle, Washington 98195
A neuron communicates over long distances (more than a few millimeters) by generating a train
of impulses which propagates down the axon to release a series of prepackaged quanta of
neurotransmitter molecules. The rate, or perhaps the patterning, of the impulse train carries the
information. One of the hallmarks of an interictal epileptogenic focus is that many of its neurons
are observed to cluster their impulses into bursts, with the intervals between impulses being
unusually short (several milliseconds). Is the bursting neuron some sort of pacemaker, driving
other normal neurons into synchronous activity and thus spreading the trouble? Or is the bursting
one observes just one of those recruited neurons, having nothing more wrong with it than an
oversized synaptic input? Or perhaps there are no pacemaker neurons; the trouble could be subtly
distributed over many neurons, changing the balance of excitation and inhibition so that the
whole circuit tends to go into a bursting-type oscillation.
There are many other ways of stating the "epileptic neuron" versus "epileptic aggregate"
dichotomy. As presented above, the argument bears a strong resemblance to an argument that has
occurred in the more general field of pattern generation: How are actions with alternating activity
and silence, such as walking or breathing, generated by groups of neurons?
While there could simply be pacemaker bursting neurons, there could also be a steady level of
excitatory drive onto two neurons (or groups of neurons) which mutually inhibit each other (33).
When A is firing, it inhibits B into silence. If one allows for inhibition which fatigues
(antifacilitation, depression), soon the declining inhibition from A will allow B to begin firing,
which will then inhibit A into silence, and so on, back and forth. Hartline (33) has reviewed the
emerging data on a number of pattern-generating circuits. Even when there are mutually
inhibitory synaptic connections which aid reciprocal bursting, the neurons themselves often have
intrinsic bursting properties too. In other words, there are redundant means of enforcing bursting;
it is not a matter of cell or circuit bursting but of both cell and circuit. While it would be more
convenient for neurophysiologists if nature would use only one bursting mechanism at a time, it
would appear that if something is worth doing, it may be done using redundant mechanisms.
In this chapter, the individual neuron is examined for its ability to exaggerate its normal output
by overproducing impulses. This emphasis on overproduction at the various stages of
computation and data transmission within the individual neuron is not to deny that
underproduction (e.g., in inhibitory neurons) could also be important. As in considering the
origins of forest fires, one can either emphasize who started the forest fire or consider the factors
enhancing the flammability of the forest. Tracing through one of the trees, i.e., the dendritic and
axonalarborization of a neuron, is useful for evaluating flammability prospects even if no one
neuron is a pacemaker.
THE PROCESSING PATH THROUGH A NEURON
Some neurons are much simpler than the neurons that we examine here. There are spikeless
neurons, such as those in the retina, where tonic transmitter release rates are controlled by the
size of a graded depolarization. Such neurons are short enough so that electrotonic current spread
suffices for communication between the postsynaptic regions, collecting information from
upstream neurons, and the presynaptic regions that output the processed result (30,31). Thus the
spikeless processing path is particularly simple.
Repetitive firing is the mechanism that allows the postsynaptic and presynaptic regions of the
neuron to be separated by more than a few millimeters; transmitter release rate is now controlled
by impulse production rate. Firing rates are in turn controlled by synaptic depolarizations, as seen
by the spike trigger zone (usually at the initial segment of the axon). Thus impulse trains can be
seen as an intermediate coding form, allowing transmitter release rate to remain proportional to
the sum of synaptic depolarizing and hyperpolarizing currents. A more detailed comparison of
spikeless and spiking modes of operation can be found in Calvin and Graubard (13).
For our purposes, we trace this processing path (Fig. 1), starting with impulse production by the
initial segment's interaction with the somadendritic region, following the impulse down the axon
through sites where the impulse might be blocked (or additional impulses created), to the
transmitter release properties of the axon terminals, and then on to the dendrites of the next
neuron to see what processes provide attenuation and augmentation of the synaptic currents
driving the impulse production processes of that neuron. At each site, we examine the features capable of overproduction, particularly those relevant to epileptic bursting.
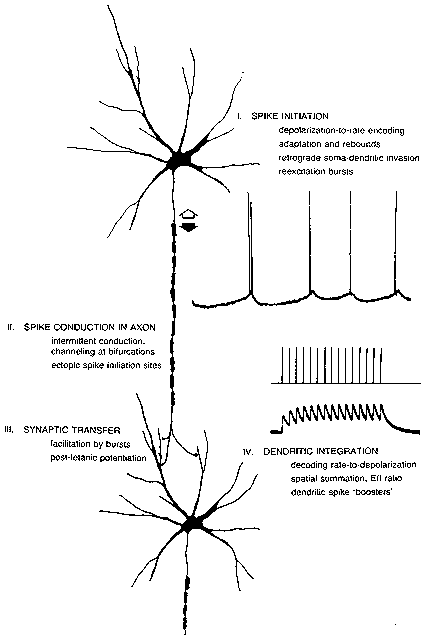
FIG. 1. The processing path from the spike initiation region of one neuron to that of
the next neuron in a chain. Repetitive firing mechanisms produce
depolarization-to-rate and adaptation features. Extra spikes (as in the double spike
in the top record) may occur from reexcitation of the spike initiation region. Spikes
may be created or destroyed in midaxon. Synaptic transmission may be sensitive to
the history of the spike train; bursts may produce increased transmitter release both
during the burst (facilitation) and afterwards (potentiation). Dendritic integration
features both temporal summation, giving a conversion of rate back into
depolarization levels, and spatial summation with its determination of the E/l ratio
and net depolarization. Special combinations of inputs may elicit local dendritic
spikes in some neurons.
Depolarization-to-Rate Conversion: Normal Rhythmic Firing from the Trigger Zone
The primary barrier to understanding the input-output conversion produced by the repetitive
firing mechanism is the confusion over the nature of the input. Traditional teaching emphasizes
the quanta!, exponentially falling shape of the unit synaptic event. It is not often recognized that
the unit postsynaptic potential (PSP), from a single impulse in a single presynaptic neuron, is
usually very small (a fraction of a millivolt, and thus no more than a few percent of the excursion
between rest and the impulse threshold). The common view, where an impulse is initiated by
several PSPs standing on the shoulder of one another, is thus misleading. The asynchronous
pitter-patter of PSPs bombarding the neuron (think of the motoneuron during a static stretch
reflex) does, however, build up a steady depolarization (7,9) analogous to the generator potential
of a sensory receptor neuron (see Fig. 5). Experimentally, one usually mimics this steady synaptic
depolarization by injecting a step of current through the recording micropipette in the soma of the
cell. A family of such steps results in a plot of current versus firing rate (the f-I curve), as in Fig.
2.
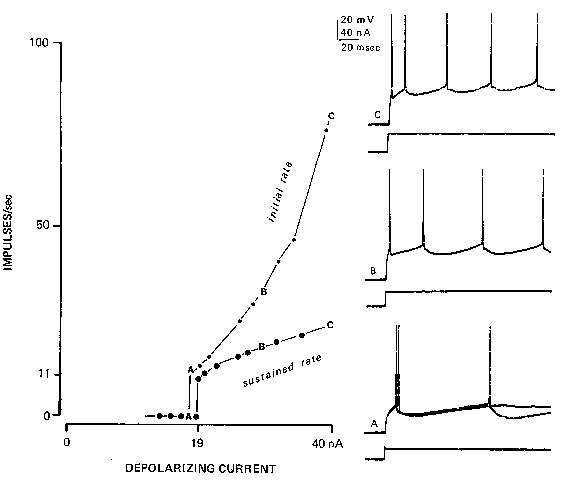
FIG. 2. Rhythmic firing in a cat spinal motoneuron, responding to steps of injected current (lower traces) injected through the recording Microelectrode into the neuron's soma, results in this depolarization-to-rate relationship (often called the f-I curv
e). Light upper line connects the points representing the initial firing rate (reciprocal of the interval between the first and second spikes of the train). Heavy lower line is the plot of the average firing rate following adaptation. At A, two responses
are superimposed; a rheobasic current gives only one spike, but a slightly larger current gives a repetitive response with an interspike interval equal to the duration of the afterhyperpolarization (90 msec) in this neuron. (Reprinted with permission from
Federation Proceedings, ref. 10.)
Essentially, there is a dead zone where subthreshold currents produce no response. Rheobasic
current usually produces only one impulse, even when the current is maintained; however,
increasing the current a little more will usually result in sustained repetitive firing for the
duration of the current. The firing rate in this situation is called the minimum rhythmic firing
rate: it is usually related to the reciprocal of the duration of the afterhyperpolarization following a
single impulse (37). Thus a neuron with a 90-msec afterhyperpolarization will usually jump from
O to l l impulses/sec as the current is increased slowly. Further increases in current level raise the
firing rate, with a proportionality constant called the sensitivity of the repetitive firing process.
This sensitivity is higher for a brief period after a sudden step in the current and then adapts
severalfold (Fig. 2).
In many neurons, the depolarization-to-rate curve rises quite linearly; i.e., the sensitivity stays
constant. For some cat spinal motoneurons, the curve has two distinct segments (37), with the
sensitivity suddenly increasing severalfold at about 30/see; phenomenologically, this change can
appear quite suddenly (54,55), although the underlying mechanisms are hardly discontinuous
(56). Of particular interest are potassium conductances in the soma (and probably the dendrites)
of a type not envisaged in Hodgkin-Huxley descriptions of axons; there are potassium
conductances activated by calcium entry (2) and those that inactivate with depolarization (21).
The latter property is especially important in generating very long interspike intervals (on the
order of seconds) with little jitter (22).
Rhythmic firing does not require these specialized ionic mechanisms; even squid axon will fire
rhythmically, although the dynamic range of its f-I curve may be small (58). The trigger zone at
the axon initial segment may, by itself, have such axon-like repetitive firing properties. However,
the impulse retrogradely invades the somadendritic region at the same time that the newly minted
impulse is propagating down the axon. This retrograde invasion activates the specialized currents
of the somadendritic region that, in turn, subtract from the synaptic currents and determine the
time taken to reach the threshold and initiate the next impulse. It has been hypothesized that the
extent of retrograde invasion is important (9,17,62). Certainly, it is likely that fairly extensive
retrograde invasion of the dendritic tree occurs in chromatolytic motoneurons (34) because of the
excitability evidenced by their orthograde dendritic spikes; their f-I curves lack the sensitivity
alterations of normal spinal motoneurons.
Extra Spikes From Normal Trigger Zones: Another Sensitivity-Changing Mechanism
Some motoneurons, while firing a spike with great regularity every 100 msec, may occasionally
produce an extra spike only a few milliseconds after a prior spike (Fig. 1, top), despite holding
the synaptic and injected current inputs to the motoneuron constant. Lowering the current slightly
may cause extra spikes to occur more often, perhaps after almost every rhythmic spike, so that
the firing is mostly in doublets. Raising the current sufficiently will eliminate the extra spikes
and restore pure rhythmic firing at the appropriate firing rate for that current. Is the doublet a
junior-sized version of an epileptic burst? Are there automatic sensitivity controls, augmenting
extra spikes if the neuron receives little input? Such questions have led to a comparative survey
of repetitive firing and extra spikes in cat spinal motoneurons (8,11,17), in neurons of cat
external cuneate nucleus and human dorsal column nuclei (15), in primary vestibular neurons of
cat (52), in cat pyramidal tract neurons (PTNs) (18,19), and in crustacean stretch receptor
neurons (14,25,29).
Extra spikes are thought to occur by reexcitation near the end of the refractory period of the prior
impulse. This requires a source of depolarizing current at the same time as the threshold drops;
usually the refractory period lasts until the membrane potential has returned to rest; i.e., there are
various wavefronts propagating down the axon, one after the other. The refractory period
wavefront never catches up with the impulse waveform. Extra spikes are thought to represent an
exception to this rule; it is not a matter of the refractory period wavefront speeding up, but rather
of the impulse waveform lasting longer so that its falling phase lags behind.
Perhaps the clearest demonstrations of reexcitation are the theoretical studies on flaring-diameter
axons (28) and the experimental investigations of lobster stretch receptor neurons using multiple
recording sites (14). In the Goldstein and Rall (28) study, a uniform axon was presumed to
change its diameter, flaring 2 to 3 diameters into a larger axon with the same membrane
properties. If there was too much flare, conduction from small-to-large axon would fail. Lesser
amounts of flare caused the impulse to broaden in duration severalfold at the junction, as the
impulse of the small axon was capacitively loaded by the increased surface area of the larger
axon. Under these circumstances, one may observe a new impulse propagating backwards from
the junction: the long duration of the impulse at the junction reexcited the small axon after its
refractory period. Thus, while the original impulse continues forwards, an extra impulse is
"reflected" backwards.
Impulses initiated at the initial segment of the axon, or antidromically propagating up the axon,
retrogradely invade the somadendritic region. Sometimes this invasion is slowed, as evidenced
by a distinct notch developing on the rising phase of the intrasomatic spike (the IS-SD break
becomes more prominent). With another recording electrode downstream on the axon, as Calvin
and Hartline (14) used with lobster stretch receptors, one may see an extra spike (Fig. 3);
evidently, the slowed invasion of the somadendritic region has allowed depolarization there to
persist long enough to reexcite the axon at the end of the axon's refractory period. The extra spike
cannot itself invade the soma retrogradely as the soma is refractory; thus, for this particular
situation, a self-perpetuating cycle cannot start.
There are other reexcitation phenomena not as readily explained by a simple two-compartment
interaction. In the typical case, extra spikes are seen to arise from the top of a depolarizing
afterpotential, which follows the prior spike. The extra spike may or may not itself retrogradely
invade the somadendritic region; if it does, it too may have a depolarizing afterpotential. In some
cases, this extra spike's aftermath may trigger still another extra spike. Such bursts of extra spikes
have been intracellularly observed in cat spinal motoneurons (8), cat PTNs (18,19), and in
various crustacean stretch receptor neurons (25,29); many of the burst discharges of hippocampal
pyramidal neurons (59) probably qualify, although their depolarizing afterpotentials are more
complex.
The postspike hump, the most prominent form of the depolarizing afterpotential, is probably of
dendritic origin. The theory of reexcitation is merely a threecompartment version of the previous
story (14). As the impulse propagates retrogradely from the initial segment trigger zone, it should
slow down as it invades the dendritic tree. Intradendritic recordings, (e.g., ref. 29), indeed show a
delayed beginning of the dendritic impulse and a broader duration (both aspects could arise just
from the cable properties of the dendritic tree, even if the spread were passive rather than active).
As the axon and soma repolarize, the dendritic spike may lag behind. This source of depolarizing
current is important. As the resistance of the soma and axon rise after their spikes, the
depolarizing current from the dendrites may cause an increasing I R product, even if the current
itself is not rising. This theory for the postspike hump (9,45) contains the necessary ingredients
for a self-reexciting process; an extra spike can trigger such a sequence as well as the first spike.
This spatial aspect of the depolarizing afterpotential is not its only aspect. The ionic mechanisms
may change from axon to soma to dendrite. One reason that the spike of the dendrite has a longer
duration may be a slow calcium current (68).
Cat PTNs are much richer in extra spike phenomena than motoneurons, probably because most
fast-conducting (> 20 m/see) PTNs have prominent postspike humps (19). More than 25~o of the
fast PTNs in the Calvin and Sypert (19) intracellular series also exhibited extra spikes during
sustained rhythmic firing driven by a step of injected current (Fig. 4). The extra spikes arose from
the top of the postspike hump (as observed from the Microelectrode site, which was probably
intrasomatic).
Historical Factors Affecting Extra Spikes
It was noted that motoneurons tend to produce extra spikes only at low rhythmic firing rates; this
was also true of primary vestibular neurons (52) and of external cuneate nucleus neurons (15).
Many lobster stretch receptor neurons fall into this category; their f-I curves may have a negative
sensitivity region where average firing rate falls as current ascends above the extra spiking
region. Other lobster stretch receptor neurons, namely those with delayed retrograde invasion
(Fig. 3), tend to produce extra spikes only at high firing rates, but this may be secondary to the
fatigue aspect in these neurons.
PTNs produce extra spikes from postspike humps only at intermediate and higher rhythmic firing
rates, but there is no suggestion of fatigue. Why they differ from most other neurons in this
aspect is unknown. The effect of extra spikes on the f-I curve is not always simple. Sometimes,
double spike firing patterns may double the sensitivity of the f-I curve; in other cases, the interval
between rhythmic spikes is lengthened by an extra spike that produces a compensatory pause. In
some cases, f-I curves may appear perfectly compensated as the firing pattern progresses from
rhythmic to doublets to triplet bursts with no change in f-I curve sensitivity. PTNs with
pronounced bursting tendencies may exhibit extra spikes atop large postspike humps even at
minimum rhythmic firing rates.
Many neurons exhibit a tendency to produce shorter interspike intervals at the beginning of a
spike train; this adaptation in firing rate seems to have a number of mechanisms in different
neurons (see list in ref. 13). About 50% of fast PTNs in the Calvin and Sypert series exhibited a
very short interspike interval of the extra spike variety after the first evoked spike, such that extra
spikes serve to augment the initial response to a sustained input. Even more interesting is the
tendency of other PTNs (11,19) and some motoneurons (8,9,17) to increase the size of the
postspike hump with successive spikes of the rhythmic response; thus an extra spike may first
occur after the second rhythmic spike of the train. Does the first spike serve to "prime" the
postspike hump mechanism so that the second rhythmic spike evokes it?
In a limited series of cells, a single spike (evoked by a very brief pulse of injected current) was
located at various times prior to a standard-sized current step evoking a repetitive response. In
these cells, the unconditioned response was a spike train with clustered extra spikes following the
second rhythmic spike. The conditioning spike shortened the interval between first and second
rhythmic spikes; at any conditioning interval shorter than several hundred milliseconds, extra
spikes would cluster after the first rhythmic spike rather than the second (11). This conditioning
effect, much longer than the duration of the afterhyperpolarization in fast PTNs, suggests that the
extra spike mechanism(s) may be primed by antecedent activity.
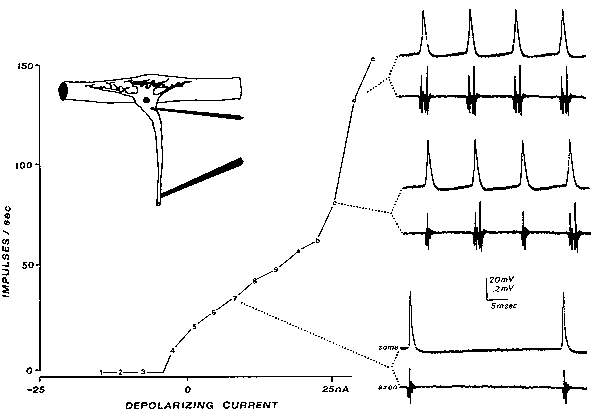
FIG. 3. Reexcitation of the axon's spike trigger zone results in extra spikes (seen
only at the axon recording electrode downstream; lower traces). Slowly increasing
current was injected through a double-barreled microelectrode into the soma of this
lobster stretch receptor neuron. With moderate heating, the retrograde invasion of
the somadendritic region is more susceptible to fatigue, slowing the invasion from
the axon trigger zone (note IS-SD notch developing in spike rising phase in some
recordings). When the soma spike lasts longer than the axon's refractory period, the
axon is reexcited. [From the Calvin and Hartline study (14), reprinted with
permission from Federation Proceedings (10).]
Extra Spikes in Pathophysiology: Augmentation of a Normal Mechanism?
The priming phenomenon noted above produces firing patterns with a cluster of extra spikes after
the second spike of the response; the first interspike interval varies with the current strength in
the usual way, but the following interspike intervals are short and relatively fixed in the extra
spike manner. This long first interval (LFI) or stereotyped afterburst pattern was first noted in
chronic monkey epileptogenic foci (20) and again in human epileptic neurons (16), suggesting
that some epileptic bursts may be clusters of extra spikes.
The short interspike intervals of epileptic bursts are indeed analogous to the typical 2-msec
interspike intervals of normal extra spike firing. The priming phenomenon also has analogous
features, helping fulfill the original hopes expressed (20) that the structured nature of the LFI
burst would place a considerable number of constraints on possible explanations. One
interpretation of LFI epileptic bursts would thus be that a moderate-sized synaptic wave sets it
off, the first interspike interval being that predicted from the f-l curve, and the afterburst being
the extra spikes that tend to appear after the second rhythmic spike. The main problem with this
interpretation is that monkey LFI bursts are seen following antidromic stimulation as well (see
Wyler and Ward, this volume), something that the cat PTNs did not exhibit. This, together with
some unusual properties of the LFI itself (20), keeps the question open as to the origins of the
epileptic LFI burst.
Assuming extra spike involvement, the high firing rates in epileptic neurons could be produced
by relatively low levels of synaptic input. The high sensitivity of extra spike repetitive firing
suggests an alternative concept to the "pacemaker" epileptic neuron. Pacemaker suggests
autogenic firing, requiring no synaptic input, but high sensitivity merely says that a small input
could give a large output.
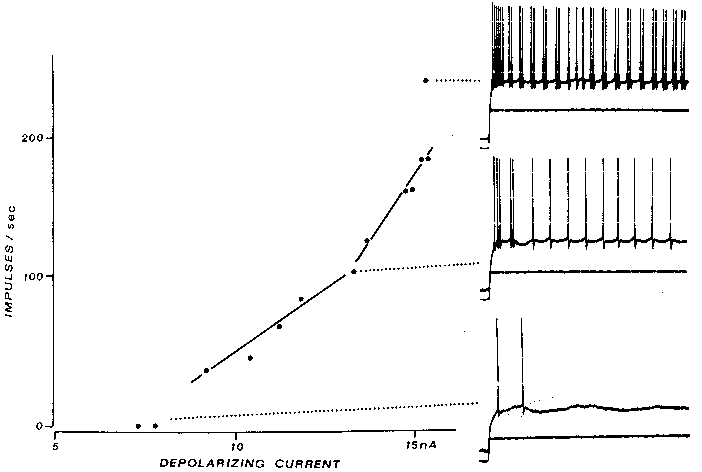
FIG. 4. Extra spikes in a cat fast PTN arise from postspike humps, not delayed
axon-tosoma invasion, as in Fig. 3. The large postspike humps characteristic of fast
PTNs are seen in the bottom trace, evoked by a near-rheobasic current; note that the
second spike's hump is much larger than that after the first spike. At intermediate
current levels, an extra spike arises from the second spike's hump (middle traces);
similar extra spikes are seen intermittently for as long as the current step is
maintained. At even higher currents, extra spikes are often seen; indeed, the
sensitivity of the f-l curve doubles in this case. Calibration bars: 20 mV 10 nA, 20
msec. [From the Calvin and Sypert (19) study. (Modified from ref. 11; copyright
1976, Raven press).]
As was noted earlier, the sensitivity of the ordinary rhythmic firing process may be controlled by
the extent of retrograde invasion, i.e., by the excitability of the dendritic tree. Extra spike
production also seems likely to involve dendritic excitability, although the duration of the
dendritic spikes and the time course of threshold recovery (8) are other important factors.
One of the major short-term ways of altering the reexcitation phenomena has been anoxia.
Niechaj and Van Harreveld (46) found that the motoneurons had increased postspike humps and
occasionally extra spikes within minutes after clamping the circulation. There are a variety of
drugs affecting crustacean stretch receptor reexcitation-type firing (65).
On a longer time scale, deafferentation is thought to affect reexcitation-type firing. In the cat
external cuneate nucleus, neurons can be partially deafferented by extensive dorsal rhizotomies
or by dorsal column sections (38). These neurons normally exhibit extra spike firing patterns
with stereotyped interspike intervals of 0.8 to 2.0 msec (15). This spontaneous activity is largely
secondary to an extensive tonic bombardment from forelimb proprioceptors. When deafferented,
the spontaneous activity disappears (38). Within a few days, spontaneous activity returns,
although it is no longer driven by forelimb receptors. The 80% of the synapses that are large and
contain round vesicles disappear; the 20% that are small, containing flattened vesicles, remain.
The extra spike-type short stereotyped interspike intervals are again prominent; in some cases,
the burst contains a dozen spikes. The bursts thus have a stereotyped appearance rather like
epileptic bursts, except that they never exhibit LFIs. This deafferentation experiment, like others
in the dorsal column nuclei (44) and elsewhere, suggests changes in the repetitive firing
properties: bursts are seen, despite presumably small (and perhaps inhibitory) synaptic inputs.
Before considering abnormal sites of repetitive spike initiation, one must ask: are extra spikes
initiated at the usual trigger zone (e.g., the axon's initial segment) or elsewhere (e.g., perhaps
starting in the dendrites and propagating down through the soma to the axon)? So far, studies of
trigger zone localization during extra spiking have been limited to crustacean stretch receptor
neurons. Extra spikes are initiated in the general vicinity of the normal trigger zone (14,25,29).
For most central nervous system (CNS) neurons, localization studies have been more difficult.
Generally, one can say that the trigger zone is downstream from the soma; but one cannot be
specific about whether it is at the initial segment, first node, and so on. Yet one can still ask
whether the sequence of retrograde invasion of the somadendritic region is the same for normal
spikes and for extra spikes. Differentiation of the spike waveforms of fast PTNs shows at least
three distinct components; all are the same in normal spikes and in extra spikes (11). This makes
it unlikely that the extra spike is beginning in the dendrites and sweeping through the soma,
opposite to the sequence of the normal retrogradely invading spikes.
Do Axons Conserve Spikes? The Creation and Destruction of Impulses at Midaxon
It is tempting to think of the axon as a rather uninteresting but reliable conduit for getting
impulses from the trigger zone to the presynaptic regions in the axon terminals. In reality, spikes
sometimes get lost along the way (48); occasionally, impulses are initiated ectopically, as in
neuralgias.
The creation of impulses at midaxon, or at other ectopic sites, is a major problem in neuralgias.
Normal dorsal root ganglion (DRG) is, unlike normal dorsal roots or peripheral nerves, tonically
mechanosensitive (36). In root or nerve, only quick distortions of an axon are capable of
initiating spikes; slower distortions may eventually block conduction without ever having
initiated an impulse (36). Yet DRG will initiate spikes tonically for many minutes after laying a
light weight atop the exposed DRG; this would appear to provide a physiological basis for the
radicular pains of herniated intervertebral discs. Focally injured roots and nerves also develop
mechanosensitivity after some days of irritation by chronic suture material; this may play an
important role in scarred nerves (such as when radicular pain to leg lifting persists after a
decompression).
There appear to be two ectopic impulse initiation processes at work. Reexcitation can occur at
the focally demyelinated regions as well as at normal DRG (35). Second, a tonic repetitive firing
mechanism develops, capable of producing sustained spike trains whenever tonic depolarizations
are present. While one ordinarily thinks of mechanosensitive generator potentials, there may also
be chemosensitivity (64). The afterdischarge seen following a priming train of impulses
conducted through a focally demyelinated region (35) is suggestive of a generator potential too,
perhaps secondary to extracellular potassium accumulation. What is different in the cases
exhibiting tonic ectopic spiking (normal DRG, demyelinated axons)?
It is interesting to consider this problem from the standpoint of specializations of the axon for
reliable conduction (12). Neural structures have a problem analogous to the impedance matching
problem in transmission lines. The active nodes must drive the capacitive and resistive load
presented by the yet-silent nodes downstream. When that load changes, as when approaching an
axon bifurcation or the unmyelinated terminal area, the requirements on the driving nodes may
be substantially increased. One finds nodes more closely spaced in a number of such situations
(12). Focal demyelination presents the midaxon with a large capacitive load, and simulations (66)
suggest that conduction may often fail unless compensatory steps are taken. Those presumed
compensatory steps may have, as a byproduct, effects on impulse initiation (as opposed to
replication) by the injured region. Could too much source conductance (too high a sodium
channel density, too little potassium conductance or leakage) make impulse origination easier, as
well as facilitating impulse conduction in the face of a capacitive load? This is the thesis
advanced elsewhere (12); central to it is the presumption that axons specialize not only for
conduction but also to avoid initiation, by positioning their source conductance in a middle
ground.
From other studies of abnormal repetitive firing, several ionic mechanisms can be mentioned.
Lower extracellular cation concentrations may initiate tonic firing (47), perhaps via shifting the
activation curve of the sodium conductance (27). Another important factor is the leakage current;
the lack of chloride currents in muscle (attributable to either lowered external C1-or to
congenitally reduced C1-conductances in myotonic goats) may also convert a faithful follower
cell into a cell with afterdischarge (1). In some situations, one may be dealing with sprouting
nerves; the sprouts are thought to be mechanosensitive, as is regenerating nerve more generally.
Ectopic Initiation in Epileptic Foci
One of our early postulates to help explain the LFI epileptic burst was that the first spike was
initiated ectopically in the axon (20). Subsequent evidence has suggested more promising
explanations for the structured bursts, but there is good evidence also for impulses initiated in
axon terminals ending within an epileptic focus. Gutnick and Prince (32) showed backfiring from
the axon terminals of thalamocortical projection neurons in a penicillin focus, and there has been
much subsequent investigation along this line (57). This suggests that antidromic impulses could
also help spread the bursting activity via axon reflex from a focus to other nondisrupted areas.
Another exception to the initiation-resistant midaxon property would appear to be axon
terminals. Normal axon terminals sometimes initiate impulses, as in the dorsal root reflex (61).
For axons in the epileptic focus, the issue becomes one of the strength of the initiating currents
(e.g., those associated with the extracellular fields of the EEG spike), the excitability of the axon
terminals for single spike initiation (as in the accommodation problem), and the repetitive firing
capability of the terminals.
Transmitter Release: Are Bursts Especially Effective?
When impulses are separated by long times, e.g., > 40 msec, the second impulse of a pair may
produce a smaller excitatory PSP (EPSP) than the first. For closer spacings, it may be larger. In
the la pathway to cat spinal motoneurons (24), the second one may be 15% larger than the first at
optimal separations.
There are, however, other synapses with more impressive facilitation properties. The
corticospinal pathway onto cervical motoneurons (49) may exhibit substantial facilitation, with
the second PSP doubling or tripling in size at optimal spacings; this is also true for the
corticorubral pathway (63). The optimal spacing is several milliseconds, much the same interval
that extra spikes prefer. This suggests that an epileptic burst might be a rather imperative
stimulus to some downstream synapses. There are longer-term effects of bursting stimuli. The
best studied is the long-term posttetanic potentiation in hippocampus, where the pathway may
remain potentiated for hours, days, and so on.
Another example of the sensitivity of a postsynaptic cell to patterning of the spike train occurs in
mammalian muscle. In single motor units of cat gastrocnemius, for example, just one short
interspike interval in an otherwise rhythmic train may double the plateau tension produced by the
train for seconds thereafter (6).
Spatial and Temporal Summation in Dendrites
Denervation supersensitivity has been one model for hyperexcitable neurons. There is some
evidence in various chronic CNS diseases for increased levels of receptors for certain
neurotransmitters (3). The ionic channels associated with extrajunctional acetylcholine (ACh)
supersensitivity in muscle are also different from junctional channels (53), and one must consider
the possibility that chronic epileptogenic foci pathology will include such altered features of the
synaptic mechanism.
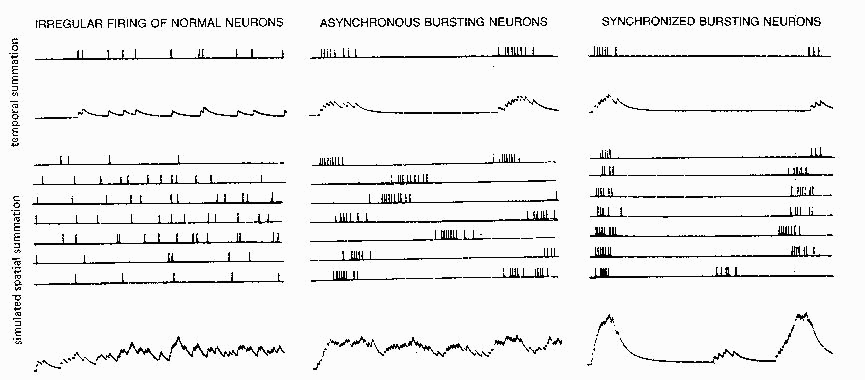
FIG. 5. Temporal summation of simulated PSPsfor an irregularly firing input (top
/efl) and an input with an epileptic bursting firing pattern (top Center) Clustering of
spikes into bursts results in peak depolarizations about three times as high as a
single PSP. Spatial summation of many asynchronous inputs (bottom left and
center) is little different for epileptic bursting inputs than for normal irregular
inputs; only when the various bursting inputs are roughly synchronized (bottom
rigby is a large depolarizing wave produced. Simulated on a cable model with
current injected (hence linear summation) using the methods of Calvin (7) and
tape-recorded firing patterns from one normal and two epileptic neurons (20).
An epileptic burst is effective in producing temporal summation of PSPs in the postsynaptic
neuron. Figure 5 (top) contrasts the temporal summation expected from an irregular spike train
from a normal cortical neuron with that expected from an epileptic bursting neuron. The mean
depolarization caused by a single input, assuming small unit PSP sizes so that the driving
potential correction may be omitted, is simply the product of the average firing rate and the area
beneath the unit PSP (9). The highest peaks of the membrane potential will correspond to the
shortest interspike intervals; since the mean depolarization level is attained in the time that it
takes a unit PSP to decay (7), epileptic bursts are easily long enough to cause depolarizations
which correspond to 500/sec average firing rates. If one averages over a longer time than one
burst, e.g., the whole sweep in Fig. 5, the mean depolarization also refers to that rate averaged
over that time.
Spatial summation of many irregular inputs (Fig. 5, lower left) gives a mean depolarization
which, assuming linear summation, is just the sum of the individual inputs' rate area products.
When bursting inputs are not synchronized (Fig. 5, center), the spatial summation gives a
sustained noisy depolarization, as in the irregular spatial summation case. When bursting inputs
are roughly synchronized (Fig. 5, right), the peaks become much higher, being predictable from
the rate area products of the individual inputs using the rates within the bursts. Nonlinear
summation effects (facilitation, driving potential decreases) will increase or reduce the net
depolarization predicted by the linear summation; however, the point still remains that bursting
in inputs producing small PSPs may not be significant until the bursting neurons are
synchronized.
The effectiveness of a synaptic input depends not only on the synaptic mechaniss but on its
location relative to spike trigger zones or presynaptic regions in the dendrites. This aspect is often
quantified by the voltage attenuation between sites in the somadendritic region; but this alone can
be misleading. Moving a synaptic input from the proximal to the distal dendrites of a model
neuron may cause only minor (10%) changes in the area beneath the EPSP 'recorded' in the soma.
The many-fold voltage attenuation between distal dendrites and soma is largely compensated by
the increase in the local size of the EPSP when situated on the high input resistance of distal
dendritic structures (31). While synaptic loci may not be especially important, in this model, for
the initiation of spikes at the initial segment trigger zone, synaptic loci may be important when
the relevant variable is the voltage generated within the dendritic tree itself. A presynaptic region
in the dendritic tree, as in dendrodendritic synapses, may be more strongly influenced by the local
synaptic inputs than by those located more proximally or on another dendrite (31). While
dendrodendritic synapses are not common in cerebral cortex, their occurrence in abnormal cortex
remains to be determined.
Dendritic Spikes
Large intradendritic voltage may also trigger dendritic spikes. By this term one does not usually
imply a propagating spike that travels down through the soma and continues past the normal
trigger zone; as noted earlier, dendritic "hot spots" usually provide a booster mechanism for
regional synaptic potentials which results in a sharp transient of several millivolts at the soma
and initial segment. Whether or not an axon spike is initiated depends on the overall integration
of many inputs, as in ordinary synaptic potential summation.
The best examples of orthograde dendritic spikes are from Purkinje cells (41) and from
chromatolytic spinal motoneurons where even one impulse in a single afferent fiber may set off a
dendritic spike (40). There is evidence that dendritic spikes have substantial calcium components
(but see ref. 51), suggesting longer duration in dendrites than soma. This has implications for
transmitter release (13) from presynaptic dendrites, for reexcitation possibilities, and for
controlling afterhyperpolarization magnitudes via calcium-activated potassium conductances (2).
In addition, calcium spikes in dendrites could have a direct effect on transmitter release from
presynaptic dendrites, e.g., by local increases in intracellular calcium concentration, as well as by
the indirect effect via membrane potential.
Besides their presynaptic effects, bursting firing patterns in the synaptic inputs could have
postsynaptic effects too; e.g., Fifkova and Van Harreveld (26) show dendritic spines that swell
following tetanic stimulation, although it is not yet known whether this is firing pattern-sensitive
or a mass action effect. If there are dendritic spikes, then input bursts might produce enough
temporal summation in the finer dendrites to cross threshold for the booster spike phenomena.
EPILEPTIC FOCI: CELL MALFUNCTION OR CIRCUIT PROBLEM?
By tracing through the mechanisms in the axonaland dendritic arborizations of an individual
neuron, the trees in the forest have been examined for their flammability prospects. In this
section, the circuit aspects are stressed. This takes two forms: the recruitment problem and the
unstable circuit problem.
Recruitment by Bursting Neurons
A bursting firing pattern in only one input to a normal neuron should have minimal effects; many
such inputs, if they were not synchronized, might produce only a steady background
depolarization (Fig. 5). A group of endogenously bursting neurons, if synchronized so that their
bursts overlap (not necessarily synchronized spike-for-spike), may recruit other normal neurons
to burst along with them by providing a large depolarizing wave of synaptic input which briefly
reaches a high level on the f-I curve of that neuron. Given typical values for unit PSP sizes,
shapes, and neuron f-I curves, it was calculated that fewer than 1% of the input of a neuron
would be required to burst synchronously to evoke a burst response (7). The other inputs could
affect the outcome by biasing the synaptic current up or down. The result merely says that turning
on such a burst pattern in 1% of the thousands of synaptic inputs could be sufficient to cause
bursting. With augmentation from facilitation or dendritic spikes, the number required would
decrease; with concomitant inhibitory bursts, more inputs would be required.
Excitatory/Inhibitory Ratios
Converting the numbers of cytologically characterized synaptic endings (e.g., round versus
flattened vesicle types) into percentages of types on an individual neuron has been done for the
cat spinal motoneuron (39) using Conradi's (23) electron microscope data. Adopting, for
illustrative convenience, the round-flat interpretation as excitatory-inhibitory (E/I), one can say
that about half of the synaptic endings on the motoneuron are excitatory (the E/I ratios are 40:60
on the soma, grading distally to 60:40 at the dendritic tips). Abnormal development can give rise
to considerable alterations in such ratios; for example, Lund and Lund (42) showed that a normal
61:39 ratio changed to 26:74 in the superior colliculus after enucleation at birth. Thus the relative
amounts of excitatory and inhibitory synaptic potentials might change with time. Ribak et al. (50)
have shown that there is a decrease in GABAergic terminals in monkey epileptic foci. Examples
of decreased inhibition exist in other chronic CNS diseases, e.g., the loss of dopaminergic
terminals in the striatum in Parkinson's disease (69).
One conclusion is that there may be cases where the flammability cannot be assessed by
examining individual trees but only by describing their admixture and specific connectivity. The
tendency of a neural circuit to go into oscillation can be described in some simple cases. In
clones, the control systems aspects of the fusimotor bias on the stretch reflex can be elucidated,
and there are thalamocortical circuits that may exhibit similar oscillatory tendencies. The central
questions are likely to be: What determines relative strengths of inputs? Is there an automatic
gain control at a synaptic level (e.g., feedback from postto pre-) or at a circuit level (e.g., turning
up the level of inhibition to produce synchronizing influences)?
FROM ANTECEDENTS TO ICTAL EVENTS
There are few theories for how a neuron changes its properties to become an epileptic neuron;
similarly, there is no comprehensive theory for how an interictal focus evolves to initiate a
seizure. In this concluding section, examples are given for both levels of theory. The purpose of
this exercise is to demonstrate the need for such theories and what ground they should be
expected to cover, not to offer serious answers.
The "Sprouting" Theory
Denervation supersensitivity stands as one of the few fundamental theories for the origin of
neuronal hyperactivity in an epileptic focus. Its virtue lies not in its congruence with
experimental facts but rather in its attempt to relate the Pathophysiology back to a more
fundamental process presumably involved with cellular development and regulation.
While the depopulation of epileptic foci and loss of dendritic spines (67) suggests denervation,
depopulation might also give rise to collateral sprouting. This may seem a paradoxical proposal,
since the most obvious feature of a focus is the truncated, weathered-looking dendritic tree. Such
shapes are also prominent in aged brains; yet careful measurements of terminal apical dendritic
branches in aged brain show sprouting (5), presumably collateral sprouting in response to the loss
or shrinkage of neighboring neurons.
If sprouting should occur in epileptic foci, what might its physiological consequences be? While
there are currently no data on alterations in the physiology of collaterally sprouting neurons, there
is information on both regenerating neurons and those undergoing normal developmental stages.
In normal development in various cell lines, originally inexcitable neurons first acquire a calcium
(Ca) spike, then a mixed sodium-calcium (Na-Ca) spike, and then most parts of the neuron make
the final transition to a sodium-only (Na) spike (60). Dendrites (see Schwartzkroin, this volume)
and axon terminals may retain mixed Na-Ca spikes. In the regeneration of a severed axon, Meiri
et al. (43) show that the cut end first seals; then, perhaps 12 hr later, the Na spike gains a Ca
component near the terminal end. This is transient, becoming undetectable with microelectrodes
in the main axon after 60 hr; by then the terminal end is bulging out, and obvious sprouting can
be seen in later days. Bray and Bunge (4) postulate a role of calcium entry in elongating the
growth cone.
This suggestion that collaterally sprouting neurons might have enhanced Ca spikes in their
dendritic trees leads one to ask what effect this might have on bursting. The most obvious
difference between Na spikes and Ca spikes is their duration, with mixed Na-Ca spikes being
intermediate in duration between the fast Na spikes and the slower Ca spikes. Thus the retrograde
invasion of the dendritic tree following spike initiation in the initial segment of the axon might
be prolonged. Because of the enhanced dendritic excitability that might occur with additional Ca
channels, the retrograde dendritic spike might be both longer and larger than in normal neurons
(see schematic spikes in Fig. 6).
It is the duration of this retrograde dendritic spike, seen at the soma or initial segment, that
creates the postspike hump that intersects the falling threshold and gives rise to extra spikes.
Thus an ordinary event initiating a spike might set off a regenerative sequence of many extra
spikes.
This theory for how bursting neurons arise begins with cell loss (secondary to anoxia, aging,
etc.), postulates collateral sprouting of adjacent dendrites, augmented calcium spikes in those
dendrites, an increased duration of retrograde dendritic spikes as a consequence, allowing the
initiation of a single spike to give rise to a regenerative burst. Since seizures give rise to
continuing degeneration in a focus (see Harris, this volume), the process might be expected to
continue in other neurons even if each neuron only went through a brief phase (as in the
regenerating axons) of sprouting and augmented Ca spikes.
While its congruence with the experimental facts may not be any more extensive at present than
the original denervation supersensitivity theory, the sprouting theory better illustrates the need to
specify each of the steps between a more fundamental cellular principle and the end product of
the Pathophysiology, in this case the interictal bursting neurons. If the end product is a seizure,
the subject is more complicated (circuits of neurons, extracellular ion changes, and so on), but a
similar sequence can be proposed to help organize the facts.
The "Epileptic Sequence" Theory
An interictal epileptic focus is sometimes thought of as a localized seizure; considerations of
extracellular potassium and calcium alterations arise along with possibilities of spreading
depression, depletion of inhibitory transmitter stores, and so forth. Yet the foregoing examination
of cell and circuit aspects of bursting suggests that collections of bursting neurons could exist
without the more elaborate environmental aspects of seizures; i.e., the focus need not be a "little
seizure." Certainly, the areas around a focus, which are recruited into a seizure, undergo a
different evolution than the focus itself. In a sense, it is like the distinction between impulse
initiation and impulse propagation; although both are impulses, the antecedent of the trigger zone
impulse is a summed synaptic potential, while the antecedent of the midaxon impulse is simply
another nearby impulse.
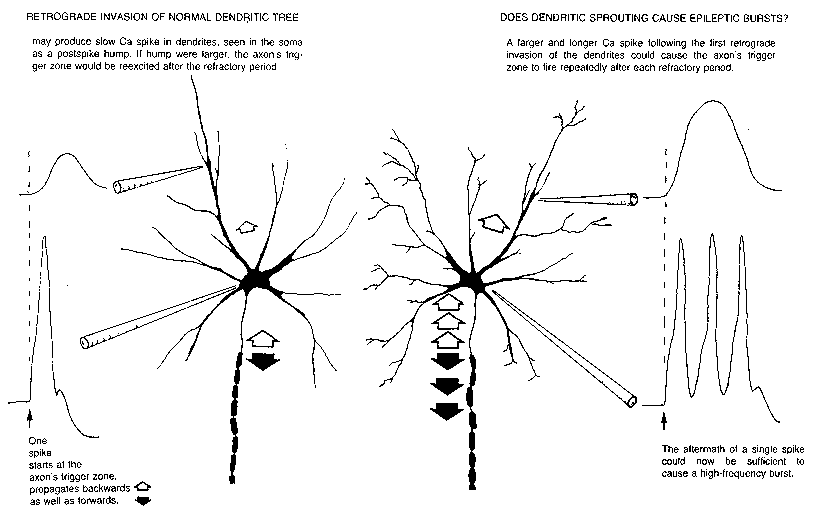
FIG. 6. Impulse initiation typically occurs at the beginning of the axon; spikes
propagate both forward (filled arrows) and backward (open arrows) into the soma
and dendrites. One explanation for the postspike hump seen intrasomatically is that
the dendritic spike is of longer duration. Ca spikes seen in dendrites might be
augmented during collateral sprouting in response to loss of neighboring neurons,
resulting in a larger and longer dendritic spike upon retrograde invasion. This could
cause repeated firing of the axon trigger zone in the pattern characteristic of
epileptic bursts.
One can propose an epileptic sequence, a set of stages through which a particular patch of cortex
evolves before and during a seizure. The items listed below are intentionally simplistic, as the
intent here is merely to illustrate the concept of an epileptic sequence rather than to propose a
particular one:
( -7) Antecedent causes (anoxia? aging? toxicity?)
( -6) Fundamental cellular mechanism responses (denervation supersensitivity? disuse responses?
sprouting?)
( -5) Dendritic excitability changes (augmented Ca spikes?) and prolonged retrograde invasion.
( -4) Reexcitation bursting triggered by normal synaptic inputs.
( -3) A progressive synchronization of previously asynchronous bursting neurons, due to synaptic
mechanisms (sleep spindles or recurrent inhibition) or field effects, leading to
( -2) Large synaptic depolarizations in normal neurons, whose depolarizationto-rate mechanism
responds with a burst. Enough synchronized neurons could now give rise to a surface BEG spike.
( -1) A decline in the afterinhibition following EEG spikes, perhaps due to inhibitory transmitter
depletion or potentiated excitation (E/I ratio increases); increases in extracellular potassium; such
declines in repolarization mechanisms could lead to a
(O) Sustained depolarization of many neurons (seizure tonic phase), followed by
(+1) Interactions between pumping mechanisms and synaptic mechanisms to produce the
instability of the clonic phase of the seizure.
(+2) Rundown of ionic gradients, depletion of transmitter stores, and their reestablishment during
postictal depression.
In the case of an afterdischarge seizure evoked by stimulating contralateral cortex, the local
epileptic sequence might start at level--2; if the seizure were spreading from the cortex next
door, it might enter the local sequence through both synaptic bombardment and diffusion of
extracellular potassium, for example. Drug-induced seizures might start the sequence by reducing
inhibition (level --1). Although it would be convenient for experimenters, it is unlikely that the
epileptic sequence actually works through a set of mechanisms seriatum; parallel actions and
interactions back and forth between levels are more likely.
Whether the paths will turn out to funnel through certain essential levels, e.g., requiring increases
in physiological E/I ratios before a seizure can start, remains to be seen. Candidates for the more
chronic aspects of epileptogenic cortex are placed nearer the top of the list; yet one could also
have a change in E/I ratios occur chronically (on the model of Parkinson's disease), which might
bypass earlier levels and have its primary effect at--1. Whether epilepsy is primarily a disorder
of a cellular mechanism or of a circuit is still unanswered; given the diversity of the epilepsies,
the answer is likely to be both. Unless there turns out to be an essential level in the epileptic
sequence which can be disabled by a specific treatment, the understanding and control of
epilepsy will depend on the elucidation of how the neuron controls its sensitivity all along the
processing path.
ACKNOWLEDGMENTS
Susan M. Johnston provided much assistance. This work was supported by the National Institutes
of Health research grants NS 04053 and NS 09677.
REFERENCES
1. Adrian, R. H., and Bryant, S. H. (1974): On the repetitive discharge in myotonic muscle fibres.
J. Physiol., 240:505-515.
2. Barrett, E. F., and Barrett, 1. N. (1976): Separation of two voltage-sensitive potassium
currents, and demonstration of a tetrodotoxin-resistant calcium current in frog motoneurones. J.
Physiol., 255:737-774.
3. Bird, E. D., Spokes, E. G., and Iverson, L. L. (1979): Brain norepinephrine and dopamine in
schizophrenia. Science, 204:93-94.
4. Bray, D., and Bunge, M. B. (1973): The growth cone in neurite extension. In: Locomotion of
Tissue Cells, Vol. 14, pp. 195-203. Ciba Foundation Symposium, London.
5. Buell, S. J., and Coleman, P. D. (1979): Dendritic growth in the aged human brain and failure
of growth in senile dementia. Science, 206:854-856.
6. Burke, R. E., Rudomin, P., and Zajac, F. D., III (1976): The effect of activation history on
tension production by individual muscle units. Rrain Res., 109:515-529.
7. Calvin, W. H. (1972): Synaptic potential summation and repetitive firing mechanisms:
Inputoutput theory for the recruitment of neurons into epileptic firing patterns. Brain Res.,
39:7194.
8. Calvin, W. H. (1974): Three modes of repetitive firing and the role of threshold time course
between spikes. Brain Res, 69:341-346.
9. Calvin, W. H. (1975): Generation of spike trains in CNS neurons. Brain Res., 84:1-22.
10. Calvin, W. H. (1978): Setting the pace and pattern of discharge: Do CNS neurons vary their
sensitivity to external inputs via their repetitive firing processes? Fed Proc., 37:2165-2170.
11. Calvin, W. H. (1978): Re-excitation in normal and abnormal repetitive firing of CNS
neurons. In: Abnormal Neuronal Discharges, edited by N. Chalazonitis and M. Boisson, pp.
49~1. Raven Press, New York.
12. Calvin, W. H. (1980): To spike or not to spike? Controlling the neuron's rhythm, preventing
the ectopic beat. In: Abnorma/ Nenes and Muscles as Impulse Generators, edited by W. Culp and
J. Ochoa. Oxford University Press.
13. Calvin, W. H., and Graubard, K. (1979): Styles of neuronal computation. In: The
Neurosciences, Fourth Study Program. edited by F. O. Schmitt and F. G. Worden, pp. 513-524.
MIT Press, Cambridge.
14. Calvin, W. H., and Hartline, D. K. (1977): Retrograde invasion of lobster stretch receptor
somata in the control of firing rate and extra spike patterning. J. Neurophysiol.. 40:10~118.
15. Calvin, W. H., and Loeser, J. D. (1975): Doublet and burst firing patterns within the dorsal
column nuclei of cat and man. Exp. Neurol., 48:406-426.
16. Calvin, W. H., Ojemann, G. A., and Ward, A. A., Jr. (1973): Human cortical neurons in
epileptogenic foci: Comparison of interictal firing patterns to those of "epileptic" neurons in
animals. Electroencephalogr. Clin. Neurophysiol., 34:337-351.
17. Calvin, W. H., and Schwindt, P. C. (1972): Steps in production of motoneuron spikes during
rhythmic firing. J. Neurophysiol., 35:297-310.
Ig. Calvin, W. H., and Sypert, G. W. (1975): Cerebral cortex neurons with extra spikes: A normal
substrate for epileptic discharges? Brain Res., 83:498-503.
19. Calvin, W. H., and Sypert, G. W. (1976): Fast and slow pyramidal tract neurons: An
intracellular analysis of their contrasting repetitive firing properties in the cat. J. Neurophysiol.,
39:420434.
20. Calvin, W. H., Sypert, G. W., and Ward, A. A., Jr. (1968): Structured timing patterns within
bursts from epileptic neurons in undrugged monkey cortex. Exp. Neurol., 21:535-549.
21. Connor, J. A., and Stevens, C. F. (1971): Prediction of repetitive firing behaviour from
voltage clamp data on an isolated neurons soma. J. Physiol., 213:31-53.
22. Connor, J. A., Walter, D., and McKown, R. (1977): Neural repetitive firing. Modifications of
the Hodgkin-Huxley axon suggested by experimental results from crustacean axons. Riophys J.,
18:81-102.
23. Conradi, S. (1969): On motoneuron synaptology in adult cats. An electron microscopic study
of the structure and location of neuronal and glial elements on cat lumbosacral motoneurons in
the normal state and after dorsal root section. Acta Physiol. Scand [SuppL], 332:1-115.
24. Curtis, D. R., and Eccles, J. C. (1960): Synaptic action during and after repetitive stimulation.
J. Physiol., 150:374-398.
25. Edwards, C., and Ottoson, D. (1958): The site of impulse initiation in a nerve cell of a
crustacean stretch receptor. J. Physiol., 143:138-148.
26. Fifkova, E., and Van Harreveld, A. (1977): Long-lasting morphological changes in dendritic
spines of dentate granular cells following stimulation of the entorhinal area. J. Neurocytol.,
6:211-230.
27. Frankenhauser, B., and Hodgkin, A. L. (1957): The action of calcium on the electrical
properties of squid axon. J. Physiol., 137:218-244.
28. Goldstein, S. S., and Rall, W. (1974): Changes of action potential shape and velocity for
changing core conductor geometry. Biophys J.. 14:731-757.
29. Grampp, W. (1966): The impulse activity in different parts of the slowly adapting stretch
receptor neuron of the lobster. Acta Physiol. Scand [SuppL], 262:3-36.
30. Graubard, K. (1978): Synaptic transmission without action potentials: Input-output properties
of a nonspiking presynaptic neuron. J. Neurophysiol., 41:1014-1025.
31. Graubard, K., and Calvin, W. H. (1979): Presynaptic dendrites: Implications of spikeless
synaptic transmission and dendritic geometry. In: The Neurosciences, Fourth Study Program,
edited by F. O. Schmitt and F. G. Worden, pp. 317-331. MIT Press, Cambridge.
32. Gutnick, M. J., and Prince, D. A. (1974): Effects of projected cortical epileptiform discharges
on neuronal activities in cat VPL. I. Interictal discharge. J. Neurophysiol., 37:1310-1327.
33. Hartline, D. K. (1978): Nervous oscillations. Conference Report 48 of the UCLA Brain
Information Service.
34. Heyer, C. B., and Llinas, R. (1977): Control of rhythmic firing in normal and axotomized cat
spinal motoneurons. J. Neurophysiol.. 40:480-488.
35. Howe, J. F., Calvin, W. H., and Loeser, J. D. (1976): Impulses reflected from dorsal root
ganglia and from focal nerve injuries. Brain Res., 116:139-144.
36. Howe, J. F., Loeser, J. D., and Calvin, W. H. (1977): Mechanosensitivity of dorsal root
ganglia and chronically injured axons: A physiological basis for the radicular pain of nerve root
compression. Pain, 3:25 41.
37. Kernell, D. (1965): The limits of firing frequency in cat lumbosacral motoneurones
possessing different time course of afterhyperpolarization. Acta Physiol. Scand, 65:87-100.
38. Kjerulf, T. D., O'Neal, J. T., Calvin, W. H., Loeser, J. D., and Westrum, L. E. (1973):
Deafferentation effects in lateral cuneate nucleus of the cat: Correlation of structural alterations
with firing pattern changes. Exp. Neurol., 39:86-102.
39. Koziol, J. A., and Tuckwell, H. C. (1978): Analysis and estimation of synaptic densities and
their spatial variation on the motoneuron surface. Brain Res, 150:617-624.
40. Kuno, M., and Llinas, R. (1970): Enhancement of synaptic transmission by dendritic
potentials in chromatolysed motoneurones of the cat. J. Physiol., 210:807-821.
41. Llinas, R., and Hess, R. (1976): Tetrodotoxin-resistant dendritic spikes in avian Purkinje
cells. Proc. Natl. Acad Sci. USA, 73:2520-2523.
42. Lund, R. D., and Lund, J. S. (1971): Synaptic adjustment after deafferentation of the superior
colliculus of the rat. Science, 171:804.
43. Meiri, H., Spira, M. E., and Parnas, I. (1980): Electrical membrane properties of a
regenerating cockroach axon. (In preparation.)
44. Millar, J., Basbaum, A. I., and Wall, P. D. (1976): Restn'.cturing of the somatotopic map and
appearance of abnormal neuronal activity in the gracile nucleus after partial deafferentation. Exp.
Neurol., 50:658-672.
45. Nelson, P. G., and Burke, R. E. (1967): Delayed depolarization in cat spinal motoneurons.
Exp. Neurol., 17:16-26.
46. Niechaj, A., and Van Harreveld, A. (1968): Effect of asphyxia on the delayed depolarization
in cat spinal motoneurons. Brain Res, 7:463-464.
47. Orchardson, R. (1978): The generation of nerve impulses in mammalian axons by changing
the concentration of the normal constituents of extracellular fluid. J. Physiol., 275:177-189.
48. Parnas, I. (1979): Propagation in nonuniform neurites: Form and function in axons. In: The
Neurosciences, Fourth Study Program, edited by F. O. Schmitt and F. G. Worden, pp. 479512.
MIT Press, Cambridge.
49. Porter, R. (1970): Early facilitation at corticomotoneuronal synapses. J. Physiol.,
207:733-746.
50. Ribak, C. E., Harris, A. B., Vaughn, J. E., and Roberts, E. (1979): Inhibitory, GABAergic
nerve terminals decrease at sites of focal epilepsy. Science, 205:211-214.
51. Roberts, W. M. (1979): Crayfish stretch receptor neurons have different sodium channels in
axons and dendrites. Neurosci Abstr., 5:295.
52. Rupert, A., Moushegian, G., and Galambos, R. (1962): Microelectrode studies of primary
vestibular neurons in cat. Exp. Neurol., 5:100-109.
53. Sakmann, B. (1978): Acetylcholine-induced ionic channels in rat skeletal muscle. Fed Proc.,
37:2654 2659.
54. Schwindt, P. C., and Calvin, W. H. (1972): Membrane-potential trajectories between spikes
underlying motoneuron firing rates. J. Neurophysiol., 35:311-325.
55. Schwindt, P. C., and Calvin, W. H. (1973): Equivalence of synaptic and injected current in
determining the membrane potential trajectory during motoneuron rhythmic firing. Brain Res.,
59:389-394.
56. Schwindt, P. C., and Crill, W. (1980): Role of a persistent inward current in motoneuron
bursting during spinal seizures. J. Neurophysiol., 43:1296-1318.
57. Scobey, R. P., and Gabor, A. J. (1975): Ectopic action-potential generation in epileptogenic
cortex. J. Neurophysiol., 38:383-394.
58. Shapiro, B. I., and Lenheer, F. K. (1972): Hodgkin-Huxley axon. Increased modulation and
linearity of response to constant current stimulus. Biophys. J., 12:1145-1158.
59. Spencer, W. A., and Kandel, E. R. (1961): Electrophysiology of hippocampal neurons. IV.
Fast prepotentials. J. Neurophysiol., 24:272-285.
60. Spitzer, N. C. (1979): lon channels in development. Ann. Rev. Neurosci., 2:363-397.
61. Toennies, J. F. (1938): Reflex discharge from the spinal cord over the dorsal roots. J.
Neurophysiol., 1:378-390.
Traub, R. D., and Llinas, R. (1977): The spatial distribution of ionic conductances in normal and
axotimized motorneurons. Neuroscience, 2:829-849.
63. Tsukahara, N. (1978): Synaptic plasticity in the red nucleus neurons. J. Physiol. fParis),
74:339345.
Wall, D., and Gutnick, M. (1974): Ongoing activity in peripheral nerves. The physiology and
pharmacology of impulses originating from a neuroma. Exp. Neurol., 43:580-593.
65. Washizu, Y. (1965): Grouped discharges of the crayfish stretch receptor neuron under
intracellular injections of drugs and ions. Comp. Biochem. Physiol., 15:535-545.
66. Waxman, S. G., and Brill, M. H. (1978): Conduction through demyelinated plaques in
multiple sclerosis: Computer simulations of facilitation by short internodes. J. Neurol.
Neurosurg. Psychiatry, 41:408-416.
67. Westrum, L. E., White, L. E., Jr., and Ward, A. A., Jr. (1964): Morphology of the
experimental epileptic focus. J. Neurosurg., 21:1033-1046.
68. Wong, R. K. S., and Prince, D. A. (1978): Participation of calcium spikes during intrinsic
burst firing in hippocampal neurons. Brain Res., 159:385-390.
69. Yahr, M. D. (1976): The Basal Ganglia. Raven Press, New York.